
Good binding data is all you need
We trained a simple model on huge amounts of in-house data and match state-of-the-art small molecule-protein binding prediction
Synopsis: Current computational small molecule binding prediction approaches rely on complex architectures, possibly to overcome a lack of data. Here, by physically generating a massive, dense dataset of millions of small molecules screened against hundreds of protein targets, we show that an extensive collection of real-world measurements enables state-of-the-art performance by a very modest model architecture. Investments in large datasets like ours - echoing Rich Sutton’s “Bitter Lesson” that general methods fueled by data and compute ultimately win - may drive performance more effectively than further architectural tinkering.
This is a pretty technical post: we go into some detail about what we built and why we think it’s great. The broad takeaway is that we’ve been arguing for years now (link) that the way to crack difficult problems like small molecule-protein binding prediction is to generate the right data, and today, we show it, because we are generating the right data. There is a clear path to getting even better at these predictions, and that path leads through the measurements we’ve been taking here at Leash.
Introduction
Recently, increased attention has been paid to the problem of predicting small molecule-protein interactions by the machine learning community. Efforts focused on pose prediction (“given that a small molecule binds a target, where does it bind within that target?”) including Isomorphic's AlphaFold3 (link), Iambic’s NeuralPlexer (link), Vant AI’s Neo-1 (link), and others. Last month’s release of Boltz-2 (link) reached beyond pose prediction and explicitly predicted the likelihood of interactions (“does this small molecule bind or not?”), promising great utility for small-molecule discovery and refinement. While a landmark achievement, the Boltz-2 binding predictions come from extensive training on public datasets, and those datasets can be stubbornly noisy and sparse despite diligent cleaning efforts.
At Leash, we have long believed that more data will be required to tackle the problem of binding prediction (link). However, existing techniques have proved insufficient to produce measurements at the quality, density, and scale required. To address this, we reenvisioned a small molecule screening process from scratch, and built extensive, automated systems to capture, classify, and interrogate many billions of physical measurements, paying special attention to data quality and density.
In the last year, we performed thousands of screens, designed and produced multiple constructs for many hundreds of proteins, and physically interrogated those proteins with at least 6.5M Leash-designed molecules each in a laboratory setting.
As a small scale proof-of-concept of the impact of our proprietary methods and datasets, we then trained Hermes, an extremely lightweight transformer architecture we created, on our data. We then used Hermes to predict binding over tens of thousands of ground-truth examples, including many quite far from our training sets.
Hermes is a fraction of the size and complexity of existing models yet competes at the state-of-the-art, and we believe its performance is due to our high-quality data. Hermes has been trained exclusively on Leash binding data and has never seen the molecules in our public data predictions. Moreover, Hermes outperforms all methods we have tried on difficult Leash-generated benchmarks. Finally, Hermes is 200-500x faster than Boltz-2.
Hermes is not a structural model: it does not predict pose or protein structure, nor does it use such features in its operations. It only predicts binding likelihood given an amino acid sequence and a SMILES representation of a small molecule. This simplicity enables its speed, and our massive data drives its predictive performance.
Results
Hermes, a simple model trained on a lot of data, extends beyond its training set
Predicting small molecule binding from amino acid sequence and a chemical formula alone is a desirable goal, one that would enable the discovery and acceleration of active chemical material for a variety of pharmaceutical, veterinary, agricultural, and industrial applications. Here, we compare models designed to make such predictions by examining those predictions on two different binding validation sets (Fig 1).

Figure 1. Left panel, model performance on novel chemical neighborhoods from proprietary Leash compounds and screening data. Right panel, model performance on public data derived from CheMBL. Leash models trained on Leash data, Boltz-2 trained on public data including CheMBL, Boltz-1 not trained on binding tasks. Hermes Papyrus performance is ensembled predictions from 3 training runs (similar to Boltz-2 binding predictions) with slightly different configurations; best individual Hermes performance is 0.669. The error bars represent the 95% confidence intervals for the weighted AUROC scores, calculated by bootstrapping over target groups.
Where our data comes from
To enable AI methods in drug discovery, we built an engine to physically collect small molecule-protein interactions at scale. We design protein constructs ourselves, produce baculoviruses encoding them, and express them in insect cell systems in small batches. We then purify those proteins with lab automation and screen them against millions of small molecules using DNA-encoded chemical library technology. Our throughput can be up to ~100 protein constructs against 6.5M molecules in a week; we have run over 2,000 individual screens in the past 12 months. Our growing protein set encompasses large numbers of disease-relevant targets: kinases, E2/E3 enzymes, transcription factors, cytokines, and more. We also mutate likely binding sites or block them with known chemical material to isolate regions of interaction between the target and small molecules.
How we determined performance on internal validation sets
To test how well these models predict, we constructed a validation set from Leash data. In brief, the Leash Private Validation Set is composed of internal data from distant chemical neighborhoods (no molecules, scaffolds, or even chemical motifs in common with training sets), but with proteins in common. The Leash Private Validation Set is 71 proteins, 7,515 small molecule binders, and 7,515 negatives, as measured on our platform. These negatives were selected to sample across similarity and dissimilarity to the positives.
How we determined performance on public data
Papyrus is a subset of ChEMBL and curated for ML purposes (link). We subsetted it further and binarized labels for binding prediction. In brief, we constructed a ~20k-sample validation set by selecting up to 125 binders per protein plus an even number of negatives for the ~100 human targets with the most binders, binarizing by mean pChEMBL (>7 as binders, <5 as non-binders), and excluding ambiguous cases to ensure high-confidence, balanced labels and protein diversity. Our subset of Papyrus, which we call the Papyrus Public Validation Set, is available here for others to use as a benchmark. It’s composed of 95 proteins, 11675 binders, and 8992 negatives.
Additional details on our internal validation and training sets will be in coming blog posts and a preprint.
Equipped with these validation sets, we predicted binding for all small molecule-protein target pairs in each set with each model. We used weighted average AUROC (link) to assess performance.
The models we evaluated
As a baseline, we use XGBoost (link), a tree-based model. XGBoost excels in low-data regimes (link), including chemistry tasks with few datapoints (link). We use XGBoost trained on Leash internal data only, Hermes trained on Leash data only, Boltz-2 (reported to be trained on public data primarily from CheMBL for affinity and PDB and distillation methods for structure, link), and Boltz-1 (reported to be trained on public data from PDB and distillation methods for structure, link). Boltz-1 is not designed to be a binding prediction model; however one of its outputs is predicted aligned error (PAE), and minimum PAE (mPAE) has been used as a proxy for binding probability (link).
We provide diagrams of the Hermes (fig 2) and Boltz-2 (fig 3) architectures below.

Figure 2. Hermes model architecture. Bottom, Boltz-2 model architecture. Hermes is a 50M parameter model built on top of two pre-trained encoder modules, ESM2 (3B) and ChemBERTa (3.4M).
Hermes is not trained on any molecules from chemical neighborhoods in the validation sets we used. Work on validation sets apart from those reported here, on Hermes trained with even larger training sets, is ongoing.
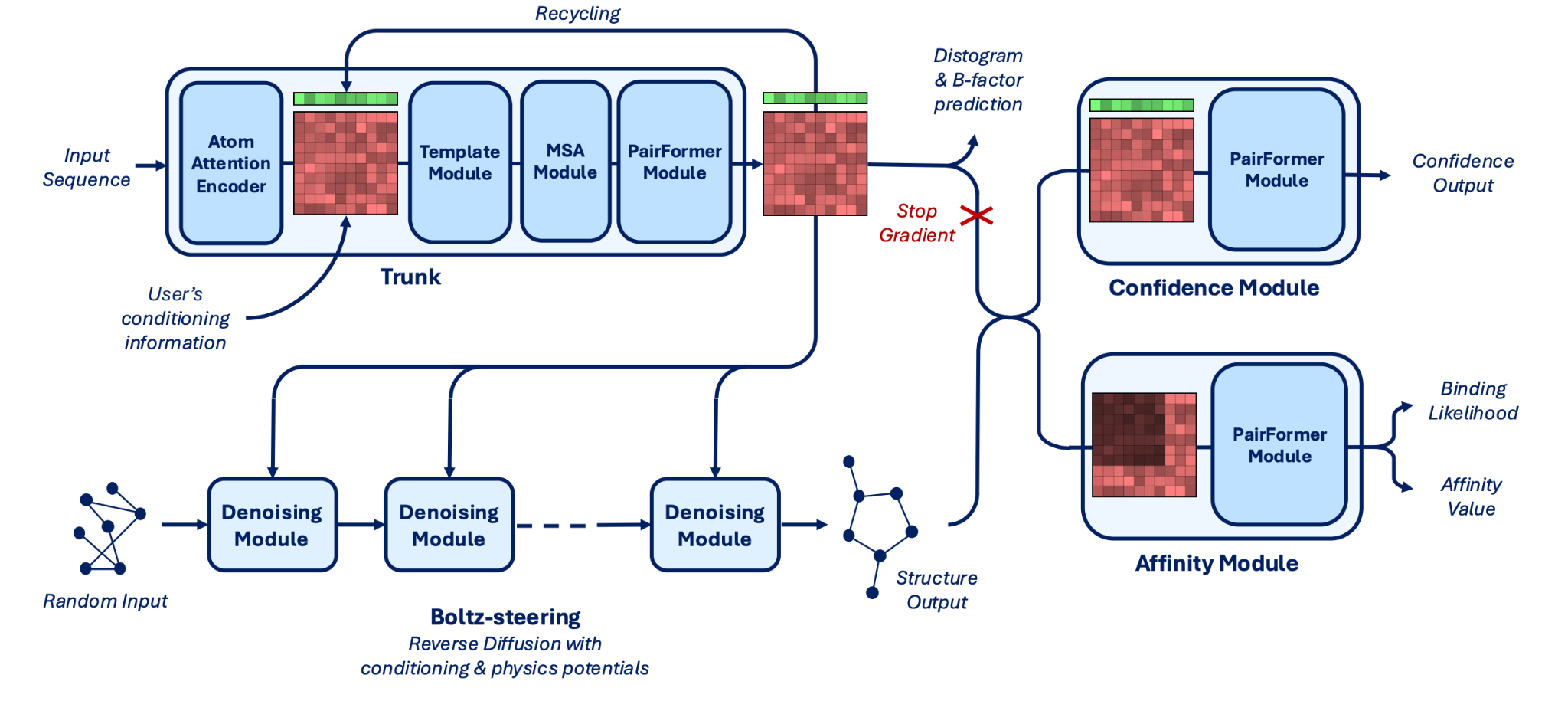
Figure 3. Boltz-2 model architecture.
Hermes can predict on difficult splits of Leash data
Our Leash Private Validation set was constructed through careful splitting and is difficult. As we removed all molecules composed of building blocks from the training set that are similar to those building blocks found in molecules in this test set, this version of the Hermes architecture was trained on substantially less data. Even so, it’s still a lot of data, and this version of Hermes shows strong performance relative to XGBoost and Boltz-2.
Hermes can predict on public data without being trained on it
When we train Hermes on even more of our data, we see strong out-of-distribution performance (on the Papyrus Public Validation Set) despite being trained on the distant chemical neighborhoods in our internal chemical library. That our internal chemical material can inform predictions on public data suggests to us that the data our platform generates contains real, translatable signal that Hermes - and future models - can exploit.
We believe that the likelihood of data leakage between our internal screens and the public Papyrus dataset is remote (see fig 4 for a comparison of distance between Leash training set molecules and public ones). Boltz-1 and Boltz-2 score similarly on the Leash validation set, but Boltz-2 is substantially better at the Papyrus validation set, perhaps because of overlap between Papyrus and the CheMBL-derived Boltz-2 binding training set.

Figure 4. Tanimoto distance of random pairs of molecules, one each from the Leash dataset and the Papyrus Public Validation Set. Distance > 0.6 is generally considered quite dissimilar (link).
A brief note on performance variability
For Boltz-2 vs. Papyrus Public Validation Set and Boltz-1 mPAE vs. Leash Private Validation Set, we evaluated Boltz models twice at 3 and 5 recycling steps. Both sets of runs were within 0.03 AUROC score of each other, with 5 recycling steps yielding better performance for each. All other Boltz scores were run with 5 recycling steps. XGBoost scores represent the best scores achieved from a parameter sweep. Hermes performance is reliable within validation sets across multiple training runs. More details on Hermes performance reliability in an upcoming preprint.
Discussion
Data make the difference
To us, these results suggest that data from our platform has the potential to make major contributions to future predictive performance, in part because we doubt the simple Hermes architecture alone could drive these results. In particular we suspect the nature of DEL chemical libraries, with their built-in SAR and repeated examples of chemical motifs in different contexts, contributed to Hermes performance. DELs are sometimes thought to be noisy assays; we find that by combining careful experimental design and comparing large numbers of screens to each other, DEL approaches are capable of providing the data scale and reproducibility driving the results we report here.
It is this scale of our experimentation that excites us most: Hermes is the worst it will ever be because our rate of training data production is likely to push Hermes’ predictive performance in the coming months and years (fig 5).

Figure 5. Leash proprietary small molecule data collection trajectory compared to public datasets. This is independent of commercial DEL library small molecule screening data that we have generated for other purposes, such as BELKA (link, link) and the upcoming STRELKA (stay tuned).
How we imagine a future of small molecule drug discovery
To find and optimize small molecules one needs to understand their likely protein targets of interest as well as off-targets that might cause harm. The combinatorial problem of searching vast chemical spaces against many protein targets simultaneously poses a challenge for the field, but recent progress gives us hope, and we demonstrate here that scaled, quality data generation methods like ours will help overcome the current limits on performance. Hermes is capable of interrogating many molecules against many protein targets with a low computational overhead, and we feel this is a direct result of our quality data generation. We look forward to using Hermes as a first-pass filter before passing promising small molecules on to more computationally-intensive, structure-based models.
In the language world, present LLMs are trained on tens of trillions of tokens. Well-trained models can then be used for few-shot or zero-shot approaches (link), like translating between languages with very few common examples (link). Similarly, we wonder if models with a solid understanding of small molecule-protein interactions could be wielded for higher-order tasks like toxicity prediction by few-shotting or fine-tuning on an experimentally tractable number of examples. While we are able to collect many billions of independent measurements of small molecule-protein pairs, we and others are unlikely to scale physical measurements of more complex systems, especially those involving whole animals or patients, to such a degree. We are hopeful that by establishing a foundational understanding of these molecular interactions, we can enable prediction at higher and higher levels of biological abstraction, leading to more rapid and effective drug design.
Hermes is named for the fleet-footed Greek messenger. Hermes was enabled by the data generation efforts of Edward Kraft, Brayden Halverson, Sarah Hugo, Mackenzie Roman, and Ben Miller; Hermes’ conception, computational infrastructure and tire-kicking was performed by Max Kleinsasser, Sean Francis-Lyon, Race Peterson, and Andrew Blevins, while Becca Levin and Julian Tu provided critical domain expertise.
 | A guest post by
|
Another issue is the _type_ of data. Can you please comment on what kind of data you are collecting? IC50s are relatively abundant in the open-source sets, but notoriously low in quality and even worse in reproducibility.
Thanks a lot! I'm glad to see this, as I've been hitting on this key for a long time now.
I see too many new models out there claiming to "beat SOTA", which bring only small, incremental improvements that more often than not fall within error bars, so, not really statistically significant at all. The methods are great, but there's a barrier we cannot seem to cross, and I've always attributed that to data, not model. No matter how much we tinker with the models, there only so much one can do with the low-quality open-source data available for most academic researchers.