
What are we able to measure?
A quick post on biological foundation models
There was a nice piece (1) on recent academic work on foundation models trained with omics data in the New York Times yesterday morning (10 March 2024). I’m focused on chemistry these days, but spent a lot of my last few gigs thinking about aspects of these problems (2, 3), so a brief walkthrough was too tempting to resist. [Edit: it was not brief.]
What is a foundation model?
It’s a model trained on a lot of data. Such a model has figured out the distributions of the problem at hand that it can start to generalize and/or generate examples of those distributions. In practical terms, this is like those models underlying chatGPT. Folks at Stanford coined the term, defining it as “any model that is trained on broad data (generally using self-supervision at scale) that can be adapted (e.g., fine-tuned) to a wide range of downstream tasks”(4).
A potentially useful part of this is that such models can be “fine-tuned”, meaning, “we trained it on a broad collection of data, and now we can fine-tune it on a very particular task with a lot less data and it can perform very well.” For example, we could take a general language model like Mistral and then fine-tune it on Tolkien to get it to write more Tolkien. Someone did exactly this and wrote it up on Reddit last month(5). I want to be clear: this Tolkien fan did it by themselves on a MacBook Pro. The effort did not require a team or even middling amounts of capital. You, reader, could do this yourself with a little chatGPTing or Googling. This is where we are now.
One can imagine a lot of uses for foundation models good at various tasks. Language models, at their core, are good at predicting what the next few words will be, given the content of the last few words.
Let’s talk about potential tasks in a biological setting. In other words:
When these guys say “biological foundation model”, what do they mean?
Here’s what the paper out of Stephen Quake’s lab (6) highlighted in the NYT article says:
[Universal Cell Embedding] is a single-cell foundation model that is built from the ground up to represent cell biology across the wide array of single-cell datasets. We envision UCE as an embedding approach that enables researchers to map any new data, including entire atlases, into an accurate, meaningful and universal space. The embedding space that emerges from UCE is highly structured and diverse and aligns cell types across tissues and species. Additionally, these cell types organize themselves in a pattern that reflects existing biological knowledge.
Ultimately, this thing they built is about taking the tiny parts of cellular machinery (proteins) - and the instructions that encode them (mRNAs) - from individual cells and putting them into a common space so that they can all be compared. They get this space by training the model on a task and then grabbing the space the model generated itself to perform the task. Such approaches can generate excellent common spaces - Recursion did something similar with images (7, look for “Image featurization for phenomic analysis”), to get a common space that was resistant to batch effects(8). In either case, you take your data after the model has been trained, convert it to the space, and then look at relationships.
An intuitive way to think about this is to imagine teaching a computer about objects from words. One could try spelling similarities as a start - the words “seat” and “chair”, while they denote very similar things, have only one letter in common. A space ruled by spelling distance would put the two of them way farther apart, and put “chair” right next to “hair”. But a different space - maybe one trained on predicting the next word in millions of sentences - might figure out that seats and chairs are semantically pretty close, just like they our in our own minds, and just like in the sentences those models observed in the training data.
Here, for the UCE paper, the task was to predict if a gene was expressed in a cell given the context of many other genes in that cell, similar to the task of predicting the next word given previous words others use in language models.
What good is a universal space?
Having a common space to put in experiments from different groups, under different conditions, and maybe from different animals, allows one to make valid comparisons across all of them. I’ve written before about the problems batch effects can bring (9), and the UCE authors claim their space is resistant to batch effects. Of course, you can’t do something like this without a ton of data, and lots of examples of the same or similar things collected across a variety of batches or conditions. For example, if we check all the genes expressed in T cells collected from patients across many labs, we can start to get a sense of how lab processing affects the data. Once we have enough of these and generate space from them - at the end of the day, T cells from healthy patients should look like T cells from other healthy patients and not muscle or neurons - we can add T cells from new labs to the space without additional model training, and the space will be resistant to those batch effects. When we get unusual examples we can trust that they are more likely to be real.
There are different ways to identify cells
To really understand this particular effort, we have to squint at the kind of data that ended up in this model. And to understand its context, we have to cover a little bit of history.
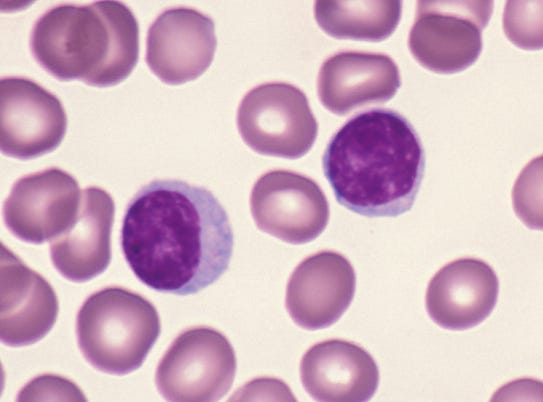
Figure 1. Red blood cells and lymphocytes. Lymphocytes are bigger/darker (10).
When cells were first described, they were described with microscopes. Human cells (and those from other critters) were categorized on whether they were obviously different-looking by the viewer. Red blood cells look different from white blood cells, and early microscopists said so (a great, brief history is here 11), but regarded some of those white blood cells, the lymphocytes, as “basically boring”. It took another half-century for biologists to realize that there were different types of lymphocytes, and they found this by discovering those different types had different functions (12). We now call them B cells and T cells, and it was decades more before researchers realized that they could use genes expressed by these cell types to discriminate them from other cell types (13). With more work and more genes we found that there are major classes of T cell subtypes, like “helper” and “cytotoxic” T cells. As we looked for more genes, we found yet more subtypes. Here’s a recent review detailing 15 kinds of cytotoxic T cell subtypes (!)(14).
More years passed and folks began to measure the expression of not just a few genes in a bulk sample but most of them (15) - or all of them (16) - with newer technologies. Crucially, these technologies measure the instructions to make the protein products of genes (mRNAs) and not the proteins themselves.
A few years after that, a group figured out how to shake organs apart into individual cells and then measure all (or most) of the mRNAs in individual cells, a technique now known as “single-cell RNAseq”. One of the first efforts did this with lung epithelium, and using basic clustering methods on the gene expression data rediscovered the five obviously prevalent cell types in that epithelium (17). I remember reading this paper when it came out and thinking, “Oh man, everyone who studies any organ is going to want to do this now to find out how many cell types there really are.” Researchers really went for it, and that’s how ~30M cells ended up in the training set for UCE.
Indeed, gene expression is what confers cell type identity (18). This is why the methods to change cell type identity (19) all involve manipulating genes that turn on other genes (called transcription factors) and not manipulating genes that affect things like cell membrane or ion transport or sugar metabolism.
This model is pretty good at identifying cells
The paper makes a point about a newly-described cell type, the Norn cell, which was discovered in, and has a functional role for, kidneys.
[UCE] could classify a cell they had never seen before as one of over 1,000 different types. One of those was the Norn cell.
“That’s remarkable, because nobody ever told the model that a Norn cell exists in the kidney,” said Jure Leskovec, a computer scientist at Stanford who trained the computers.
This is neat. What happened here is that UCE got a pretty good fingerprint for a Norn cell or a cell very much like it hiding in the training data, and when newer datasets from other organ types were put in that embedding space, it found Norn cells in kidneys.
This nearly-Norn cell is probably from the lung; at least, that’s my prediction. Turns out there are a lot of shared cell types and developmental mechanisms between kidneys and lungs (and frog skin!). A cell type my colleagues and I described in frog skin looks very much like cells found in the kidney (20), and at the time I predicted those cells would also be found in lung, since frog skin looks a lot like lung epithelium. I looked with 2009 technology and didn’t find them, but folks did by 2019 (21).
So, to sum up, the UCE presumably found a cell type in its training data and noticed that cell type in a new organ.
… but identifying cells doesn’t exactly tell us what they’re doing
Both the NYT piece and the UCE authors say big things like
U.C.E. also taught itself some important things about how the cells develop from a single fertilized egg. For example, U.C.E. recognized that all the cells in the body can be grouped according to which of three layers they came from in the early embryo.
“It essentially rediscovered developmental biology,” said Stephen Quake, a biophysicist at Stanford who helped develop U.C.E.
Do cell types from the same germ layer express some similar sets of genes? Yes! (22) They also look different under a microscope, which is why germ layers were first described two centuries ago (23). UCE took all those gene expression profiles and found them to be closer to each other when they share the same germ layer origin, which is intuitive and makes sense. Principal Component Analysis also can also do this (figure 2). PCA is a simple mathematical transform invented in 1901 and not some fancy neural net that anybody would say “discovered” something on its own.
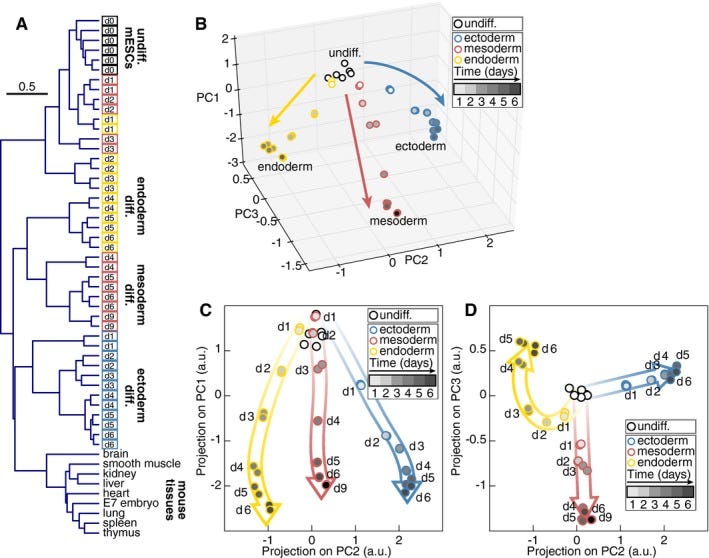
Fig 2. Global gene expression changes in germ layer specification. From (22).
Those genes in those three germ layers are different because of upstream events that themselves took a century to uncover and rely on secreted proteins (which we can’t measure with mRNAs) and embryo shape (which we very much can’t measure with mRNAs)(24). Put another way, we can see which way the dominoes fell, but with mRNAs it’s more difficult to identify the finger that pushed them. (Here is an inside baseball account of how that finger specifying germ layers was identified 25). Turns out developmental biology is very complicated. UCE does not understand it, not by a lot.
This is still really cool
I’ve written before about how data quality and batch effects are critically important (9), and having a space to reduce those and compare experiments across lots of groups is very useful, especially for a measurement modality that is so scalable. To call UCE a “foundation model” sort of implies that it can transfer what it’s learned to other tasks - like if it discovered a deeper structure in cell gene expression behaviors that’s more obscure to us - and it’s not yet clear to me that we’re there yet, though.
What is biology?
ML works best in modalities where we can make a ton of measurements. Right now in the life sciences, those measurements are imaging and DNA sequencing, and we can use DNA sequencing to measure mRNA quite well. But biology is a lot more than that: it’s protein abundance, protein translation rates, protein degradation rates, protein modifications, protein localization, protein complexes, organelle structure, cell shape, cell stiffness, lipids, local concentrations of metabolites, local concentrations of gases, ion transport, electrical charge, cell-cell interactions, tissue-level interactions, and on and on.
We can only measure a few of these to the level that ML really needs. In the near term, ML will get really quite good at derivatives of those measurements. Things like cell type for mRNA measurements, for example. But mRNA changes are detectable in about a third of active biological perturbants (26). Imaging can detect more but still, it’s only about half (27). There is so much of biology we can’t detect in amounts enough to help with ML approaches, and we need laboratory methods to collect those different data types at scale. Time to go work on that problem!