
BELKA results suggest computers can memorize, but not create, drugs
We bet machines will create novel druglike material in the future, but probably not now
“The best way to discover a new drug is to start with an old one.”
– 1988 Nobel Prize in Medicine winner James Black
At Leash, we try to discover drugs and improve the process of drug discovery. One way we think drug discovery will be more efficient in the future is by leveraging modern machine learning methods - not unlike those good at text generation you might use via chatGPT - to help in identifying and designing druglike material.
We’re not the only team to believe this! There are many groups out there trying to teach computers biology (I wrote about some of that work here) or to teach computers target-based drug discovery (finding a protein or small molecule to bind a specific, disease-causing protein target). At Leash, we believe that by showing computers what small molecules binding to protein targets looks like - across many protein targets and many small molecules - computers can learn enough about their properties to predict the likelihood of novel molecules binding. This could make drug discovery and development go a lot faster. We want to be able to design, and not just get lucky.
There aren’t a lot of examples of small molecules binding to proteins in the public domain and the existing ones are pretty biased (I wrote about that here), and so we built a lab to physically measure new ones ourselves. Once you have the examples, though, there are many ways to train computers with them, far more than we had bandwidth to try on our own. We were also mad that there weren’t many public examples out there already.

To address these issues, we launched the Big Encoded Library for Chemical Assessment (BELKA), a Kaggle competition where we provided more data on this problem than previously existed in the public domain all put together - 300M examples aggregated from 4B physical measurements of small molecules binding (or not) to three different protein targets - and challenged the machine learning community to use these examples to teach computers to predict if novel, unknown small molecules might bind to those three targets. Roughly 2,000 teams of machine learning enthusiasts competed over the course of three months.
We are here to report that this staggering amount of data is simply not enough for computational methods to predict novel bioactive chemical material reliably. We don’t think anyone is capable of generalizing to new chemical space yet.
What is the problem exactly?
The problem is to accurately predict if a given small molecule binds to specific protein targets.
Machine learning people create and evaluate their models by taking their data and splitting it: they have their models learn on some of the data (the “train” part of the split) and then have those models predict on the rest to see how they do (the “test” part of the split). Having very similar molecules in your test-train split gives your models opportunities to memorize building blocks rather than really learn the underlying behavior of those chemistries, which is not what you want. (Other groups have noticed.) We don’t want models that can only do this for some molecules, like those memorized, and not others - we want a general model, one that can perform well on small molecules that look nothing like each other.
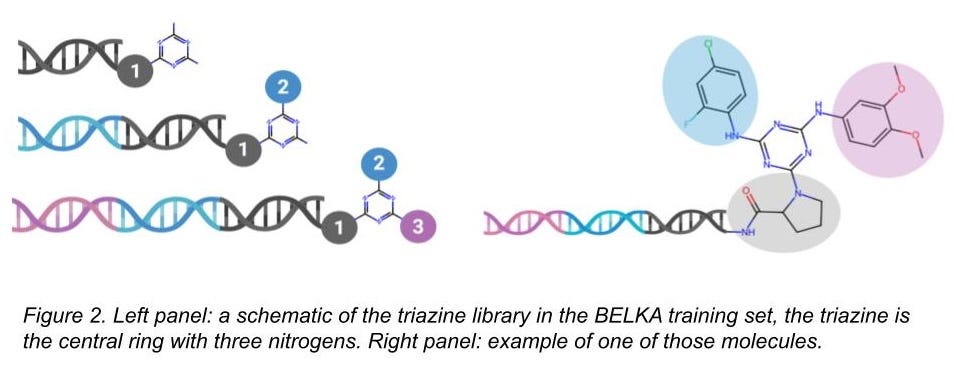
For BELKA, we provided binding data on about 133M small molecules, and every single one of these molecules had the same chemical core (a triazine). Our chemical libraries are DNA-encoded libraries (DELs), and they are manufactured by adding chemical building blocks at a few different positions on the triazine core (Figures 2 and 3). The library has a total of 305 building blocks at the first position, 768 at the second, and 951 at the third. (Sidebar: attentive readers will note that 305*768*951 = 223M, but there are a lot of examples where the same building blocks could be at either positions 2 or 3, and that means an identical molecule was made. We don’t count that twice after aggregating.)
We took this DEL and screened three protein targets with it (BRD4, EPHX2/SEH, and ALB/HSA), identifying which of those molecules bound and which ones did not. We provided the data for Kaggle competitors to train their models and held out a subset of it for them to evaluate those models (that test-train split part we described above). We also deviously threw in some very different chemical material as an extra challenge.
How can we tell if anyone is any good at this?
We challenged their models to predict if small molecules bind and checked how they did.
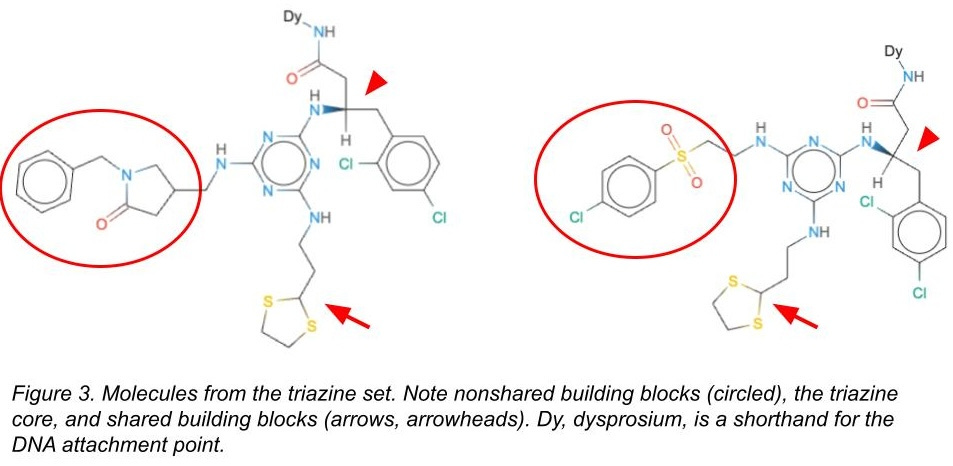
To see how good various teams were, and to prevent pure memorization, we created our test-train split carefully.
There are some building blocks that are in both the test and the train sets (see Figure 3). We’d expect models would memorize these and do a great job predicting behavior on molecules in the test set containing those building blocks. But memorizing is cheating! It won’t help you generalize to new building blocks or new molecules.
There are some building blocks that are only in the train set or only in the test set. We’d expect models to have a more difficult time with these. Still, the molecules in both the test and train sets would have the same triazine core, and so maybe the models can still learn some useful things somewhat easily.
For the test set only, we also added a completely different chemical library with different cores and different attachment chemistries that we had also screened against these targets (Figure 4). It’s a subset of a chemical library we designed here at Leash we call kinase0. Kinase0 is so different that only models capable of generalizing have a prayer of accurately predicting binders from this library.
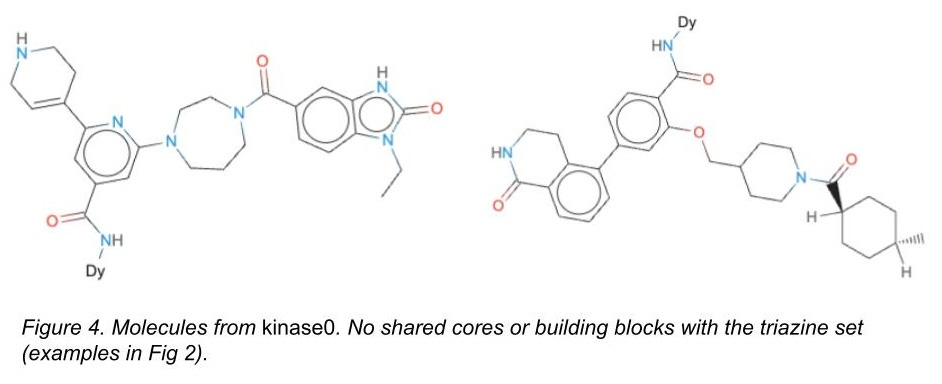
With these splits in hand, we asked competitors to train on the triazine set, and then asked them to predict the likelihood of binders from a) a lot of triazine-core molecules with building blocks shared between test and training sets, b) a lot of triazine-core molecules that did not share building blocks with the training set, and c) a lot of kinase0 molecules, which shared nothing at all. We asked them to demonstrate memorization, to demonstrate generalization within molecules that had the same core, and then to demonstrate generalization into new chemical space entirely.
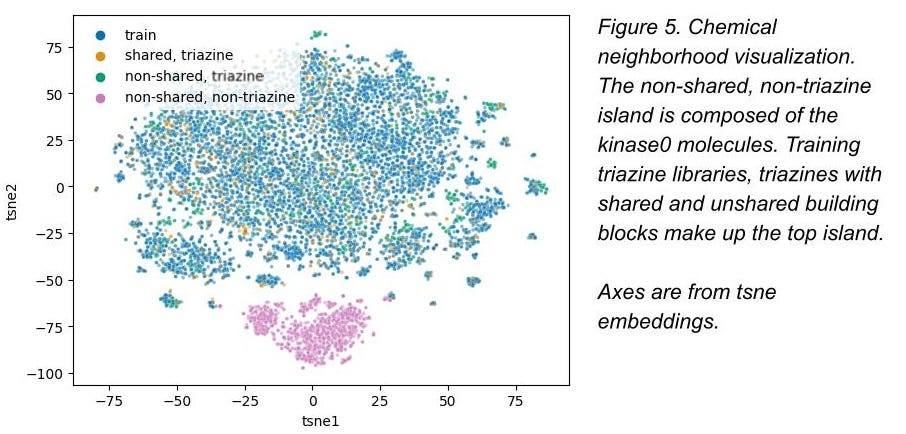
A good visualization of the central problem here is in Figure 5: the training set and non-shared triazine core molecules are all in that upper blob of chemical space, but kinase0 is entirely in the lower pink blob (plot from Kaggler Hideaki Ogasawara; source here). Kinase0 doesn’t look anything like those triazine molecules, but we asked the Kagglers to predict its properties anyway. That’s what this fancy computer stuff is supposed to help us do!
Why do we want to generalize, anyway?
We want to be able to design drugs and not just get lucky or rely on massive, expensive search.
Was anyone any good at this?
No.
As I mentioned, we split the test set into three groups, and we challenged the competitors to make predictions on potential binders to three different proteins. Here, we use precision-recall curves to demonstrate performance (click here for a precision-recall refresher if you need it).
In figures 6 and 7, we show the top competitor performance for the test set. You can see the winner (link) does pretty good with shared building blocks - ones models could memorize. He does mostly worse with non-shared building blocks (not as memorizable but still sharing a common triazine core), and can’t predict in new chemical space at all. All of the competitors looked like this or worse.
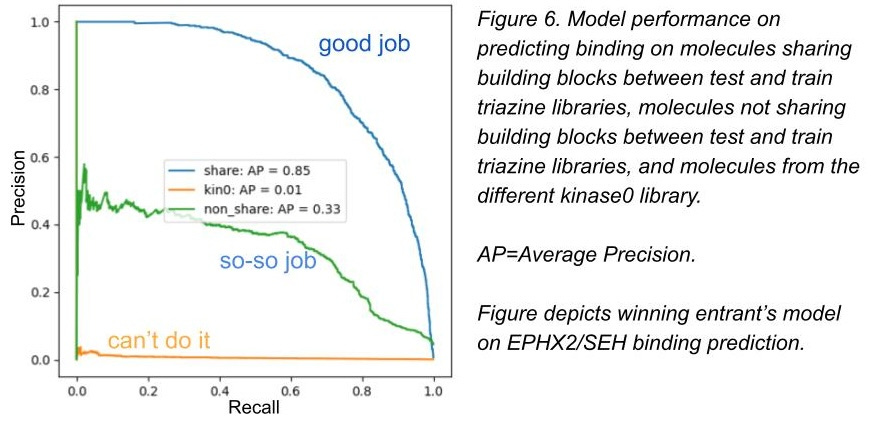
Three of the winners in the top 5 all used the same tutorial notebook with minimal changes, which suggests the randomness in rerunning the same simple, but noisy, notebook a bunch of times performed better than nearly all of the extremely diligent and fancy approaches lots of other competitors tried.
Predicting molecule binding in novel chemical space, even with literally hundreds of millions of training examples, is very very difficult.
Does this get at the practical heart of the problem?
Not exactly, but we simulated the practical heart afterwards. No one could do that, either.
Ultimately, we’re looking for new drugs. Rather than being good at predicting all of these molecules, what if we just restricted ourselves to the most confident predictions? While we felt this wasn’t a great metric to evaluate contestants in the Kaggle competition, it did seem worthwhile to have a look at how the winners might have fared.
We imagined the following scenario: one could run a DEL screen, train on the data, and then use that model to predict the likelihood of binding from a big orderable catalog - like the Enamine catalog - and run a smaller number of the top-predicted molecules on more expensive functional tests. This is a very common strategy; a different machine learning chemistry contest, the CACHE Challenge, operates this way. Google and X-Chem tried it too.
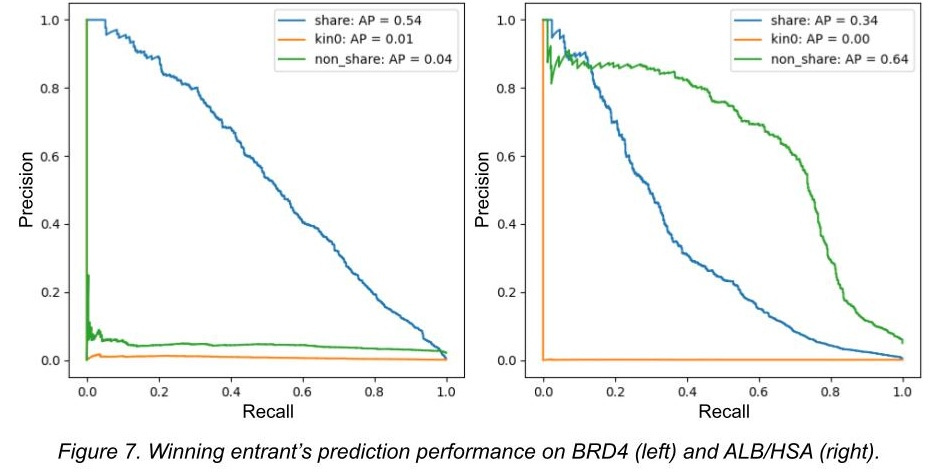
To simulate this scenario, we asked how many true binders from the top 100 binding predictions on kinase0 each competitor made for each target, pretending that kinase0 was an orderable collection. The contest winner predicted 2 true binders out of their top 100 most-confident binder predictions for EPHX2/SEH. For BRD4 and ALB/HSA, none of their most-confident predictions actually bound. There were ~120k kinase0 molecules in our Kaggle competition and ~1000 hits per protein. If you were just guessing, you’d expect to get roughly 1 out of 100 for each target.
Other contest winners performed similarly: their success rate was within the range of picking random molecules out of kinase0. If they were using this approach to identify new chemical material for, say, a drug program, they would do no better than throwing darts at a catalog.
Why does this look worse than other efforts?
We’re not sure, but BELKA tested a lot more molecules than any other public assessment so far.
There have been many attempts to predict active small molecules with computers and test them physically for activity. One recent study (in what was surely a massive amount of work and coordination) put together results for some 318 protein targets from many research groups, finding a hit rate of roughly 5-8%, across many protein target classes by ordering predicted molecules from commercial sources. This recent paper on a single target reports a 60% hit rate. We do not see anything near this level of enrichment, even with 2000 participating teams and targets with half a million repeatable binders per target, hundreds of millions of negative examples, and hundreds of thousands of test molecules.
This discrepancy is the subject of further study here at Leash, but we have some ideas. Many reported ML-generated successes might stem from building block or chemical motif memorization, as compared to generalization and out-of-distribution predictions. Something similar might be said for virtual screening that might memorize similar pockets across related proteins (e.g., kinase ATP pockets). Docking might be better but difficult to scale (the one docking effort we know of in BELKA did not work well at all). Existing orderable chemical space itself is heavily biased towards biologically-active chemical motifs, increasing the likelihood that random orders might produce actives, which could make catalog-picking a more successful exercise than predicting binders from our DELs.
There’s more. Other efforts often have a final step where compounds are selected by a human medicinal chemist; we didn’t do that here. Physical testing of hits can involve leaky functional assays or high compound concentrations (>10 µM) that could be more nonspecific, whereas the concentrations of individual compounds in our DEL screens is quite low, in the femtomolar or even attomolar range. DELs may be hitting multiple target pockets, and perhaps the models learned one pocket from the triazine set and kinase0 hit elsewhere. Maybe building block diversity is more important than molecule diversity, and these DELs only have a few thousand building blocks. We provided an enormous amount of data for only three targets; those three could be lousy choices for molecule generalization (gPCRs in particular yield high hit rates from docking approaches, see the discussion section here). Maybe DELs as a screening modality have some fundamental flaw that torpedoed these efforts. Other possibilities may elude our imagination.
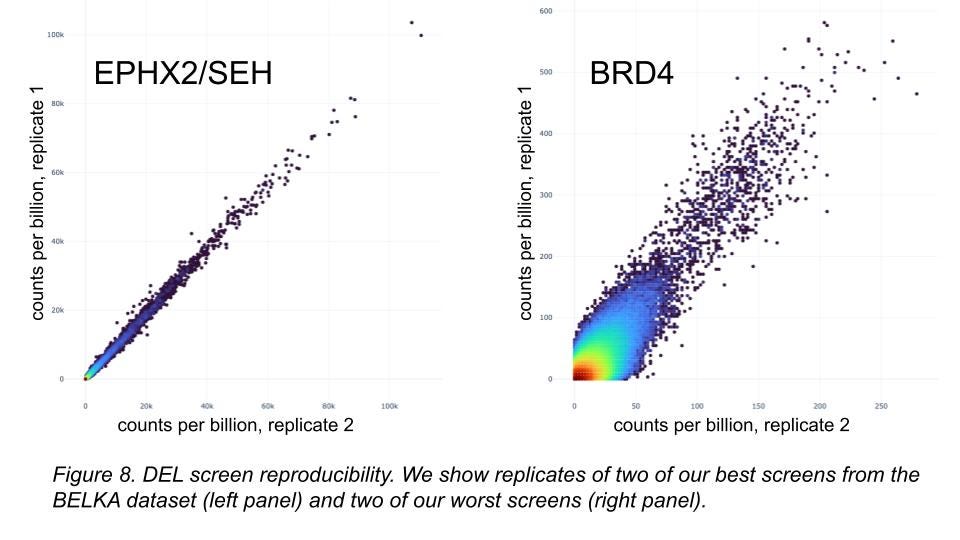
Finally, we could be bad at lab experimentation ourselves (Figure 8). Maybe our dataset is terrible! To help test that idea, we’ll be releasing the raw data from all screen replicates and controls, about 4B physical measurements, very soon (September 2024, on Polaris). We invite the community to examine their quality when we do, and perhaps use them in the future to show generalization with superior methods.
The takeaway
Predicting protein target binders in new chemical space is very difficult.
We released more data than currently exists in the public domain, with minimal batch effects (all collected by the same team at the same time in the same facility with the same reagents and processed with the same pipeline), and 2000 teams of machine learning researchers used it to try to generalize into new chemical neighborhoods. Despite these efforts, we saw essentially no success. A computational chemist contestant put it well in the Kaggle forums:
This result is troubling to me as this is exactly what computational chemistry and cheminformatics is supposed to do - translate to previously unseen structures. While there is certainly a benefit and enrichment for the triazine non-shared BBs, the non-triazine set shows no enrichment for hits based on predictions. It is an interesting result and certainly an interesting problem that remains to be solved.
We too believe the field has a long way to go before generalization is on the table. Like other domains solved with machine learning, such as chess or Go or identifying objects in images, we believe more examples of the thing we’re trying to model, across its distribution, is the surest path forward. For now, though, we think computers can only do what Nobel laureate James Black recommended: find new drugs by memorizing the old.