


How to screen a lot
DNA-encoded chemical libraries plus small batch protein synthesis are an underappreciated combo for measurements at scale

Here at Leash, we think ML/AI approaches work best when they’re shown large numbers of examples of the thing those models are trying to predict (ref 1). “Large numbers” here has some looseness here but to set your mind right, current LLMs are typically trained on tens of trillions of tokens (ref 2). Given that we’re not-bad at protein folding now (not everyone agrees on this, ref 3), it’s instructive to note that protein folding models are mostly built on UniRef sequences, which number in the hundreds of millions (ref 4).
There’s no such large collection of small molecules binding to protein targets. There are some smaller ones like bindingdb (ref 5) or its bigger cousin PubChem (ref 6), but really nothing on the scale of what was needed to crack language or protein folding. A lot of this is because measuring small molecule interactions at scale is itself hard. You have to make the molecule, store it in a tube, expose it to your protein target one at a time, and have some sort of readout to see if it did something. Lots of labor!
Enter DNA-encoded chemical libraries, or DELs (more detail in ref 7). They’re chemical libraries in a pool (thousands to billions of molecules in a single tube) that have DNA barcodes covalently attached. You dunk your protein in the tube, fish it out, and sequence the DNA of chemical binders. In the life sciences at this particular moment, there is no more scalable measurement technology than DNA sequencing.
At Leash, we want to essentially be the Google of small molecule behaviors in a protein context: that means we will collect and catalog many millions of molecules against every protein target. It’s a big job, and we think it wouldn’t be possible to even try without DELs and existing DNA sequencing technologies.
Illumina DNA sequencers changed everything
When Illumina instruments came online in the mid-2000s, a bunch of different folks - your Joe Eckers, your Bing Rens, your Steve Quakes, your John Stamatoyannopouloses, your Steve Henikoffs - suddenly had cheap access to sequencers and cheap things to try given existing molecular biology knowledge, and in a decade or so they collectively produced this amazing creative fluorescence of techniques and protocols (RNAseq ref 8, methyl-seq ref 8, ChIPseq ref 9, Hi-C ref 10, DNAse-seq ref 11, single-cell RNAseq ref 12, etc). Several us here at Leash were around to witness, or contribute, to this fluorescence.
Given the scale of what DNA sequencers can measure, we felt it only natural to leverage that scale to measure small molecule-protein interactions in the numbers we’d need for real ML/AI solutions. So we took the genomics methods development ethos and applied it to DEL screens. For ML/AI purposes, though, we also felt it was really critical to measure not only a ton of small molecules, but their interactions with a ton of proteins. That meant we had to figure out how to screen large numbers of them from my basement, and after that, a shopping mall in Utah.
We do DELS different from everybody else
DEL screening is historically pretty slow (see this CRO saying it takes 3-6 months after the protein is obtained, read this joke about it on BlueSky). Because it’s perceived as such a big time/money investment, folks will want to be very certain that their DEL screen is likely to work, and so before dunking their favorite protein in the chemical library they’ll put a lot of effort into making sure that protein is perfect. They’ll grow a huge vat of it, they’ll purify it many times, they’ll run activity assays on the protein lot to make sure it’s functional.
We don’t do that here. We screen really fast, and we screen a lot. Right now we screen 100 homemade protein constructs a week with a lab team of 4, there’s no way we can put deep effort into characterizing those proteins extensively. To be very clear: we manufacture the proteins here and screen them fresh, ourselves, at a scale nobody else can match. When we go out in the world and talk about it, folks used to big vats sometimes think we’re generating garbage data. How could being so fast and loose possibly work?
Read on.
You can break a screen in predictable ways to identify if the protein has (probably) folded correctly, and you can also compare it to other ones to help
The next few sections in this post are going to get a little technical, and if that’s not for you, I wanted to TLDR them up front. Proteins, at their hearts, are little machines that work because of the shapes they form, and they make those shapes by folding a single long peptide chain into various loops and sheets and so on (AlphaFold won the Nobel Prize this year for its ability to predict those shapes by looking at that single long peptide chain alone). If the proteins are folded correctly, they’ll do their jobs; if they’re misfolded, they won’t. Inside an egg are all the ingredients to make a chicken. But if you boil that egg (which misfolds the proteins!), you are probably not going to get a chicken out of it afterwards. Same goes with the individual proteins in our screens. Most DEL, or general biochemistry, groups run functional tests on their own proteins, which usually takes a lot of material and adds a lot of time to the experiment.
We don’t run functional tests on our proteins (e.g., we don’t test if they can actually pull a phosphate off ATP), so we had to come up with creative ways to estimate if they folded correctly. One way to do this is by examining the screens themselves: you’re profiling these things with millions of chemical probes, why not use those profiles? If you know a protein has a pocket your library binds to, and you break that pocket and the binders go away, doesn’t that suggest the unbroken pocket folded correctly? Below, we describe the various ways in which we use our screens to determine if known pockets are present and folded correctly, along with other strategies. Let’s dive in.
Screening with known competitors suggests active protein
Many proteins have described pockets and there are known inhibitors or binders of those sites. We often screen with and without such inhibitors, inferring that the active sites have folded correctly if a somewhat selective blocker can occupy them and displace DEL hits (figure 1). These inhibitors might be chemical; for DNA-binding proteins we block with consensus motif oligos, which then outcompete similar motifs in the DEL barcodes. Even if we don’t get any DEL hits, rediscovering consensus motifs suggests that the DNA-binding proteins folded correctly (ref 13).
[A quick note on these figures: we have a tool where we can circle hits in a scatterplot and then locate them in a different scatterplot. Inside the loop they are dark black, and outside the loop they’re a lighter grey; when we see them in a different plot, the circled hits are red.]
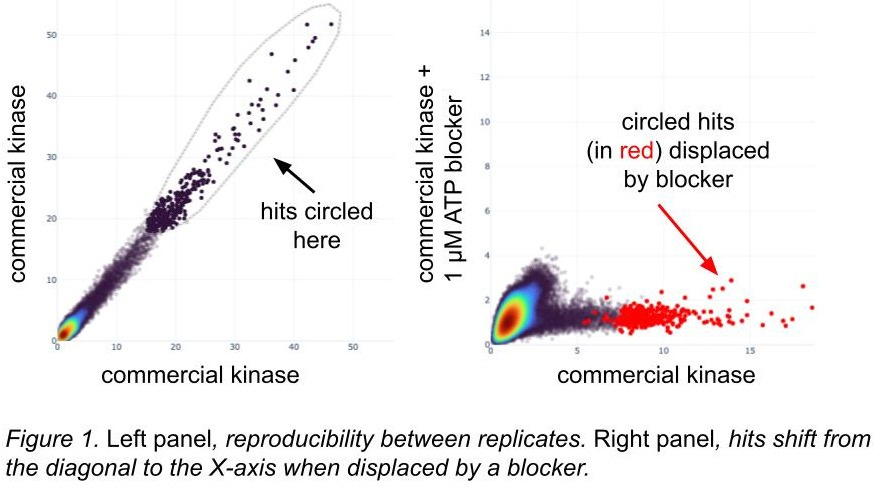
Screening with known disabling mutations suggests active protein
For proteins with known inactivating mutations, we’ll engineer them and screen them. Like the use of blockers, we infer that known structural variants likely to kick out active binders from our libraries will give us information about the differences we see in hits between wild-types and mutations (figure 2): if the broken ATP pocket displaces hits, that’s some evidence that the wild-type one is folded correctly. We feel even better about it if the hits displaced by the ATP mutant are the same hits as those displaced by the blocker known to occupy the wild-type ATP pocket, and in figure 2, they are! The overlap is imperfect, but also our homemade kinase has different domain boundaries and a few other tweaks. We capture these amino acid differences and show them to our models while training.
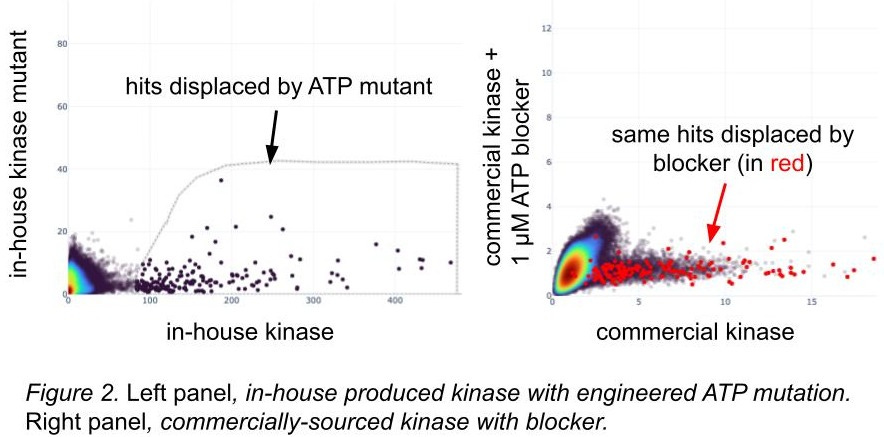
Screening with known phosphomimetic mutations suggests active protein
Additional structural knowledge allows us to engineer protein variants to validate our methods. For some transcription factors, phosphorylation of key residues introduce conformational changes allowing for additional complex members to bind, and phosphomimetic residues (amino acids that look like other phosphorylated ones, ref 14) can do a pretty good job of simulating this. We employ this trick pretty frequently: in figure 3 you can see the complex member dislodging DEL hits, and those hits are only present when the phosphorylation event is present.
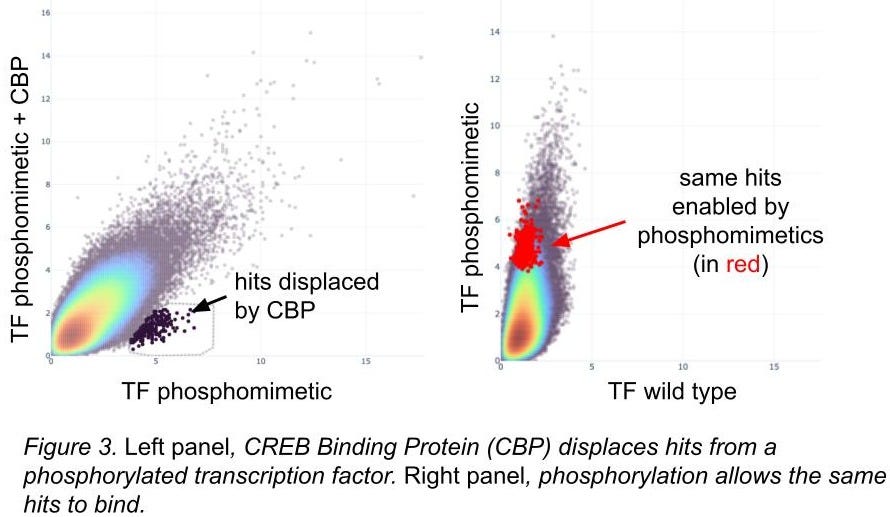
Phosphomimetic residues don’t always work; we focus on ones with strong structural evidence of efficacy. For cases like this we’re happy with the results.
In-house proteins get same hits as commercial proteins that have been tested for activity
To test overall similarity between screens, one metric we use is SubSet Similarity (sss). It allows us to quantify screening result comparisons. An sss of 0 indicates no hits shared between two screens; an sss of 1 indicates all hits from at least one of the two screens are shared in the other. Here are a handful of proteins where we’ve screened commercial versions that have protein lot-matched functional activity data against proteins we manufactured onsite, and the sss between them.
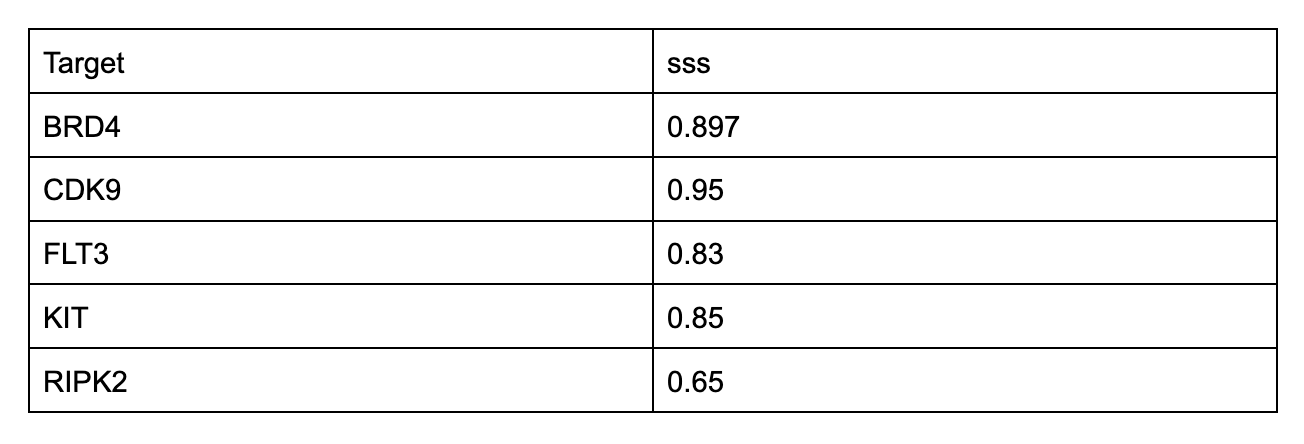
We don’t make such comparisons for tons of proteins - sourcing them can be difficult, slow, and expensive - but we get results like this when we do.
Gross similarity between family members suggests active protein
We have built apps to compare all screens to all other screens in the same chemical space (figure 4). Along several similarity metrics (including sss), obvious protein family and subfamily relationships are recapitulated from the hits in our screens. The relationships between these families and subfamilies would be difficult to create by accident; we take their rediscovery as further support that those proteins are folded correctly. By contrast, we regard distant outliers with deep suspicion.
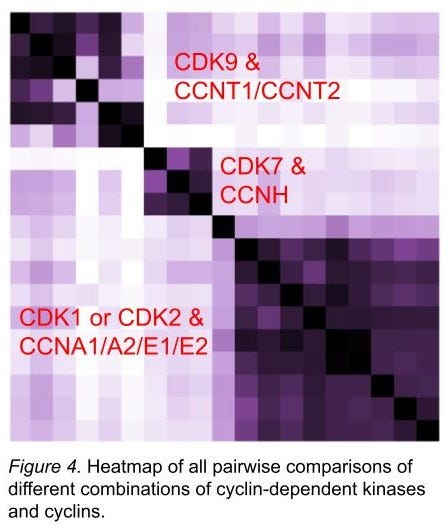
In figure 4, we show that different combinations of cyclins and CDK9 look like each other, whereas it’s more difficult to parse between CDK1 and CDK2. Inhibiting CDK2 while sparing CDK1 has proved challenging (ref 15), so we are encouraged to see that they’re both quite similar in our screens, too. (This doesn’t mean we can’t find small numbers of hits that are selective, though!) We extended this exercise to many CDKs and cyclins - many dozens of combinations - and have found lots of interesting stuff on selectivity and the role of which cyclin is present in determining what sort of chemical material binds. And that’s just the CDKs. As of today (April 2 2025) we’re at about 400 proteins (at least 2 constructs each) or complexes with hits, and each protein construct or complex has been screened against at least 6.5M molecules.
Putting it all together
Overall, by anchoring our proteins to function with demonstrably functional proteins from commercial sources, blockers, and mutations and then comparing those anchors to all other proteins screened to evaluate their relationships, we have a pretty solid basis on which to evaluate if a particular protein was produced and screened successfully and whether the data is suitable for downstream applications. We use the assay itself and smart experimental design to characterize our proteins. Under these conditions, we still fail about 35% of our screens.
Sounds cool and all, but what’s your off-DNA functional validation rate?
We get asked this, too. Across a number of targets but at a much smaller scale (these tests are expensive), our functional validation rate is better than 90%.
Thanks for reading! This is the beginning of a series of posts where Leash team members will be discussing what we’ve been building over here and some of the choices we made - even for the screening part alone, we left a lot of those choices out of this post. If the prospect of screening every protein against many millions of molecules is enticing to you, please reach out!